
The U.S. Food and Drug Administration (FDA) is approving more drugs with the help of real-world evidence (RWE) than ever before. Between 1995 and 1997, 19.4 percent of the FDA’s approvals came from having one adequate and well-controlled study plus confirmatory evidence, such as RWE. Between 2015 and 2017, that figure jumped to 47.2 percent.
The FDA has signaled cautious optimism for the appropriate use of RWE in support of FDA decisions, such as in its 2001 guidance on choice of control group, and in its framework on RWE. In 2019, the FDA approved three products—PRETOMANID, Ga 68 DOTATOC, and ZOLGENSMA® (onasemnogene abeparvovec-xioi)—in which an externally controlled trial provided the evidence needed to meet the “substantial evidence” standard.
In this article, we examine three recent and important cases of RWE use in establishing substantial evidence. Among these three examples, RWE enabled the FDA to approve treatments for serious conditions:
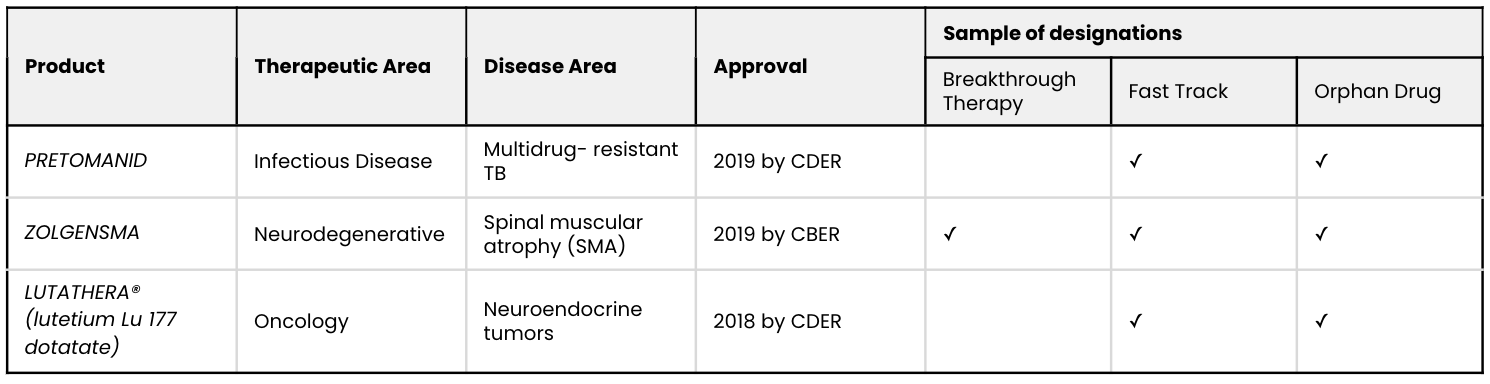
Source: FDA drug approval documents.
Overview of the RWE studies submitted for regulatory approval:
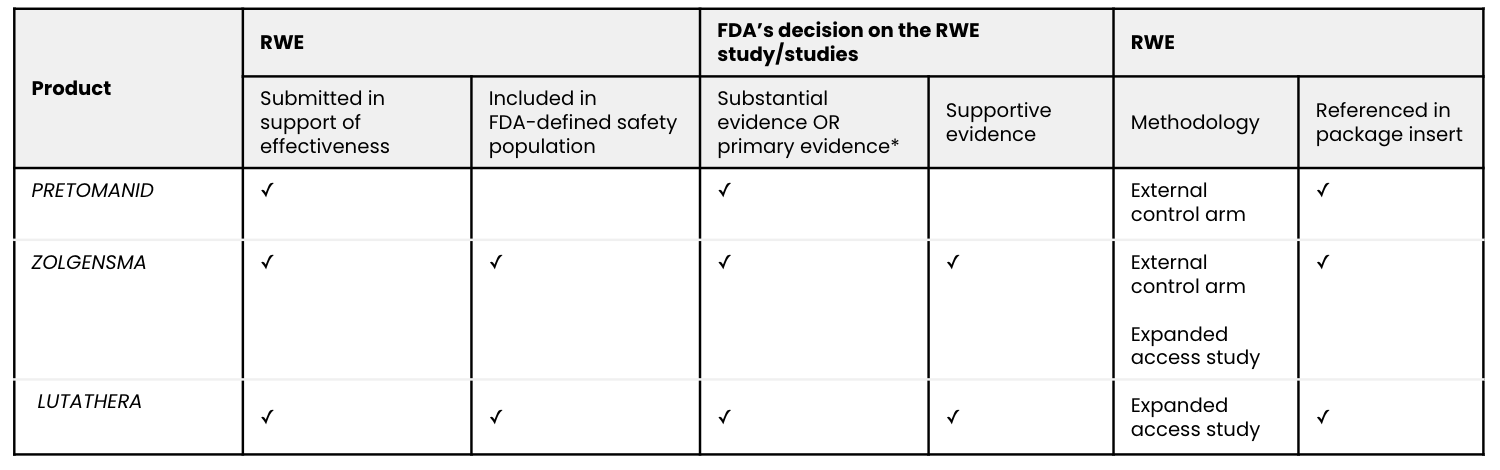
Source: Aetion analysis; FDA drug approval documents.
EXAMPLE 1: PRETOMANID for the treatment of highly treatment-resistant tuberculosis
On August 14, 2019, the FDA approved TB Alliance’s pretomanid tablets as part of a combination regimen with bedaquiline and linezolid for the treatment of people with a specific type of highly treatment-resistant tuberculosis (TB) of the lungs.
Key findings from the FDA’s Multi-Discipline Review and Other Review(s):
The safety and effectiveness was primarily demonstrated in a phase 3, open-label, noncomparative trial, Nix-TB (NCT02333799; n=109), of a three-drug regimen of pretomanid, bedaquiline, and linezolid. The applicant provided substantial evidence of effectiveness by comparing its single phase 3 clinical trial to historical controls. Of the 107 patients who were evaluated six months after the end of therapy, 89 percent showed treatment success, meaning the absence of bacteriologic failure, relapse, or clinical failure. This significantly exceeded the 50 percent treatment success rate in prespecified historical controls.
Support for use of an external control arm in this study:

Source: Aetion Science analysis in collaboration with select advisors; FDA-review documents; FDA guidance on demonstrating substantial evidence of effectiveness.
EXAMPLE 2: ZOLGENSMA for the treatment of spinal muscular atrophy
On May 24, 2019, the FDA approved AveXis’s ZOLGENSMA® (onasemnogene abeparvovec-xioi), “for the treatment of pediatric patients less than two years of age with a specific type of spinal muscular atrophy (SMA).”
Key findings from the FDA’s Summary Basis for Regulatory Action and the BLA Clinical Review Memorandum:
Comparison of the results of the ongoing phase 3 trial, CL-303 (NCT03306277; n=21), to available natural history data of infants with SMA provides primary evidence of the effectiveness of onasemnogene abeparvovec-xioi. Results of the phase 1 trial, CL-101 (NCT02122952; n=15), additionally supported effectiveness. Other potential sources of effectiveness data, such as the U.S. expanded access program, are ongoing and therefore incomplete. Contributions to the safety database include: the phase 1 and 3 trials, other ongoing trials, and patients treated through the U.S. expanded access program.
Support for use of an external control arm in this study:

EXAMPLE 3: LUTATHERA for the treatment of neuroendocrine tumors of the gut
On January 26, 2018, the FDA granted approval to Advanced Accelerator Applications USA, Inc. (AAA)’s LUTATHERA® (lutetium Lu 177 dotatate) for “treatment of adult patients with somatostatin receptor (SSTR) positive gastroenteropancreatic neuroendocrine tumors (GEP-NETs) including foregut, midgut, and hindgut neuroendocrine tumors.”
Key findings from the FDA’s Multi-Discipline Review:
A randomized controlled trial, Netter-1 AAA-III-01 (NCT01578239; n=231), provided the primary safety and efficacy evidence. But a single-arm study based on an expanded access program, called the Erasmus Medical Center (EMC) trial, “provided supportive data on safety” and “support for the indication for the treatment of patients with GEP-NETs other than those arising in the midgut.”
Support for use of an expanded access program source:

Source: Aetion Science analysis in collaboration with select advisors; FDA-review documents; FDA guidance and overview on expanded access.
Industry-wide shared learning process
These three cases illustrate the promise of RWE going forward. As former FDA Commissioner Dr. Rob Califf offered: “Properly done, RWE will vastly expand knowledge about medical products throughout the life cycle of the products.”
Aetion is systematically assessing approval documents for drugs and biologics to understand when, where, and how RWE studies are used in applications. This work allows us to learn from the growing empirical evidence base, and in anticipation of the FDA’s September 2021 draft guidance on RWE and its March 2023 revised guidance on RWE.
As the FDA implements its plan to develop draft guidance on RWE, we invite you to learn along with us on the FDA’s decisions.